
Most teenagers do not typically face the risk of death when interacting with others on a daily basis. However, this is the harsh reality for nineteen-year-old Ally Nichting. When she was four months old, Ally’s parents took her to Duke Hospital after they had consistently noticed abnormal digestive activity. It was there that doctors delivered the life-changing news and diagnosed Ally with cystic fibrosis.
Cystic fibrosis (CF) is the mutation of the cystic fibrosis transmembrane-conductance regulator (CFTR) protein, which is located on the surface of the cell membrane. The CFTR protein allows for the passage of chloride in and out of the cell. However, because CF patients have mutated CFTR proteins, they end up with a thick build-up of mucus in their lungs, which is just one, and arguably the most prominent, of the many issues associated with cystic fibrosis.
The dense mucus that builds up in the lungs of CF patients causes them to be more prone to bacteria that grows very easily in that environment, leading to infections in the lungs. CF patients must be very cautious about interacting with one another due to the risk of cross contamination. If two CF patients interact in person, it increases the risk of developing infections because of the different bacteria in each person’s lungs. In most cases, this could potentially cause long-term, severe illness or death. The Cystic Fibrosis Foundation recommends that individuals with the disease stay at least 6 feet away from each other to reduce the risk of exposure to germs that become airborne with a cough or sneeze. Even a simple seasonal cold can send CF patients to the hospital for two weeks or more.
In addition to build-up in the lungs, mucus can also build-up in other areas, such as the pancreas. The pancreas is important for the release of enzymes that break down food. According to the Cystic Fibrosis Foundation, nearly 90 percent of CF patients experience mucous blockages in their pancreas, thus creating a need for enzyme supplements, to allow food to be properly digested.
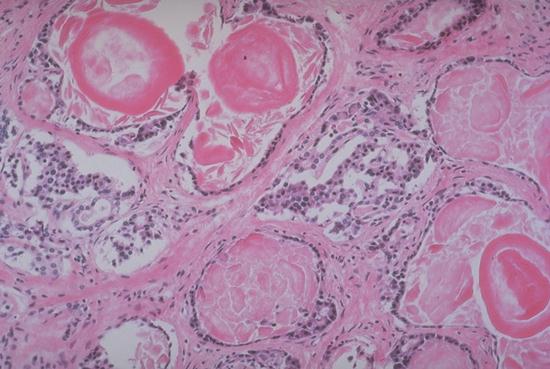
Pancreas sells secreting abnormal amounts of mucus
The majority of CF patients end up having mutations within their already mutated CFTR protein. The most common mutation being the F508del mutation, which is the deletion of the 508th amino acid in their CFTR protein sequence. Some patients, like Ally, have more than one mutation, which prohibits her from taking certain medications to treat her various symptoms.
Thankfully, in the past four months, the FDA approved a new cystic fibrosis triple drug treatment, Trikafta, that works for 90% of patients with cystic fibrosis gene mutations, including Ally’s specific mutation. When she got the news, Ally was elated to find out that there was a drug that accommodates patients like her. “A few years ago, a big drug came out that was very promising but it was only able to help certain mutations, and I had one of those mutations but the other one I have is extremely rare so I wasn’t able to be on it. Seeing that this drug helps 90 percent of the CF gene mutation thrills me!” said Ally.
Trikafta is administered orally via pill and delivers three drugs: tezacaftor, ivacaftor, and elexacaftor. Tezacaftor and elexacaftor correct the CFTR protein so they are able to achieve proper placement on the cell membrane’s surface. Ivacaftor is a potentiator that facilitates the opening of the CFTR protein to allow chloride to pass through.
In a clinical trial comparing Trikafta (tezacaftor, elexacaftor, ivacaftor) to a double drug treatment Semdeko (tezacaftor, ivacaftor), patients were seen to have more clinically robust benefits with the triple drug treatment versus the double drug treatment. Patients on the double drug treatment had a 10-percentage point increase on their pulmonary functions test (PFTs), specifically on the forced expiratory volume in one second (FEV1), whereas patients on the triple drug treatment had a 17.4-percentage point increase in their FEV1.
In addition to increased FEV1 function, doctors also reported that patients had improved sweat chloride concentrations, showing that chloride was able to pass through the CFTR proteins more efficiently. Patients also discovered improved health related quality of life (HRQoL) on Trikafta versus the double drug treatment.
Because there are only about 30,000 cases of cystic fibrosis in the United States, the disease often goes unnoticed or people don’t realize the full extent to which cystic fibrosis affects everyday life, whether it be being able to breathe or just digest food. “CF affects literally everything, more than people realize. Sometimes I forget the amount it affects,” said Ally. Something as simple as the human touch is taken for granted by so many humans. If Ally comes into contact with the wrong person’s bacteria, she could face extreme health consequences or even death, which is hard to think about as a nineteen-year-old.
The development of Trikafta is important because it is able to treat a broad range of cystic fibrosis symptoms to an extent that has not been treatable previously. Without awareness, fundraising, and financial support, the development of Trikafta would not have been possible and Ally would not have this new weapon to fight her battle with Cystic Fibrosis.
S. Ung
References:
Heijerman, Harry G M, et al. “Efficacy and Safety of the Elexacaftor plus Tezacaftor plus Ivacaftor Combination Regimen in People with Cystic Fibrosis Homozygous for the F508del Mutation: a Double-Blind, Randomized, Phase 3 Trial.” The Lancet, vol. 394, no. 10212, Nov. 2019, pp. 1940–1948., doi:10.1016/s0140-6736(19)32597-8.
Kwong, Eugenie, et al. “The Impact of Cystic Fibrosis-Related Diabetes on Health-Related Quality of Life.” Journal of Cystic Fibrosis, vol. 18, no. 5, Sept. 2019, pp. 734–736., doi:10.1016/j.jcf.2019.03.007.
Mahase, Elisabeth. “Cystic Fibrosis: Triple Therapy Shows Promising Results.” Bmj, 2019, p. l6347., doi:10.1136/bmj.l6347.
N/a. “Cystic Fibrosis Foundation.” Cystic Fibrosis Foundation, 0AD, www.cff.org/.
Voelker, Rebecca. “Patients With Cystic Fibrosis Have New Triple-Drug Combination.” Jama, vol. 322, no. 21, 2019, p. 2068., doi:10.1001/jama.2019.19351.
Image Credits:
Microscope pancreas image: http://www.pathology.washington.edu/content/page-img/359/spa-img0034__small.jpg
{kind=link}